| Contents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
MoleculeMolecules interact with each other to build pathways. A molecule is anything that is subject to reactions. Most molecules have a mass, be it a small molecule like ATP, or a protein. No difference is made between receptors, enzymes, second messengers, transcription factors or other special kinds of proteins. A molecule can also be a group of such entities, like a protein family, a state of such an entity, like the phosphorylated form or a complex of several other molecules. And finally a molecule can be part of another molecule, either non-covalently bound as in a complex, or covalently bound such as ubiquitin-like proteins. The reason for such a wide scope for this class is to catch anything that shows specific signaling behavior. A molecule can serve in four fundamental roles in the context of a reaction: it can be an educt, a product, an enzyme or a modulator. Enzyme, educt and modulator are the inputs of the reactions, while products are the outputs. For semantic reactions molecules are only grouped into signal donors, which are the inputs, and signal acceptors, which are the outputs. We use two other collections, modified forms and complexes, to keep the states/modified forms and complexes separated from the family grouping hierarchy.
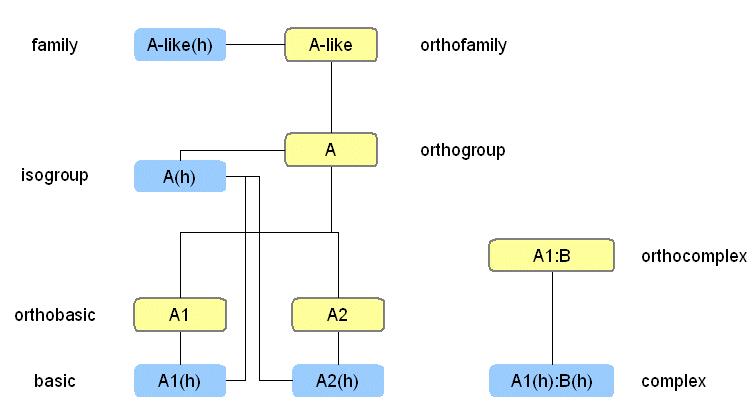
Orthofamily/FamilyGroupingImportant attributes of all components are the collections that allow expression of family relationships. Each component can act as a group of other components or as a member in such a group. In the case of molecule, this grouping represents family relationships between the molecules. In the case of reaction, this grouping can be used to assign reactions to a general class of reactions, as it is done successfully in the EC nomenclature. The third possibility is the use of mixed groups, which contain both reactions and molecules, and correspond to nets or explicitly stated pathways. This is interesting since it would support the appealing idea of building networks from subnets in a modular fashion. Grouping molecules into families is essential for a usable signal transduction database. When the family relationships are stated in a formal way, it is easy to write algorithms that exploit them in user queries, while marking inherited properties as inferred information. The prefix "ortho" is used when the family entry is not specific for a certain species or higher taxon (Fig. 1). The family relation is implemented by the groups/members collections.
Orthogroup/IsogroupFor a single gene, different isoforms such as splice variants may exist. Sometimes in the literature a signaling activity is first attributed to a single molecule, and later it is discovered that there is a whole group of similar molecules. Therefore, a special type of molecule entry is needed, which we term "isogroup" for taxon-specific entries and "orthogroup" for orthologous, non-species-specific ones. To these abstracted group entries all the information can be assigned, where it is not known which specific isoform is involved. Orthobasic/BasicMolecules of the type "basic" contain data for a specific isoform, e.g. splice variant, to which an amino acid sequence can be assigned. Again, the prefix "ortho" is used to generalize information for orthologous isoforms from different species. Figure 1 sums up all the various hierarchical grouping relations that molecules in the database can have.
Classification methodsSince it is easier to examine the sequence of a gene or protein than to investigate its function, a certain behavior is usually shown in detail for only few members of a protein family, and homologs are added to the functional group by sequence or structural similarity. Based on this assumption, various good databases exist that try to classify proteins and map sequence motifs to functional annotation. They cluster proteins by multiple sequence alignments and use common structural motifs, "profile" patterns, or Hidden Markov Models derived from these alignments to classify new proteins. Sometimes these methods can correctly predict the function, but sometimes not. Thus it is common practice to group molecules into families on the basis of sequence similarities, even if they do not share common behavior. For a signaling database, it is advantageous to group molecules that show common signaling behavior, that is, to group them by function. Since it is the function we are interested in, we would like to group only by function. On the other hand, we would like to draw as much as possible upon expert knowledge, and stay coherent with the groupings that already exist. To solve this dilemma, the best option seems to be that we group the molecules as it is done traditionally, but link signaling only to those molecules for which it has been shown. Given the hierarchical tree for in Fig. 1, if the reaction has been demonstrated for A(h), we link that reaction to A(h) only. We link statements made on a generalized level, like the ones from review papers, to nodes on a higher level in the molecule hierarchy. Given the tree we can link the general activation of A-like proteins to the orthofamily entry "A-like". The context of the original literature statement is preserved.
States (modified forms)The unmodified form of a protein and all its modified forms are its states, where the modification can be by covalent binding, by complexation, or by change of environment. The protein per se is a concept that is based on the observation that there is only one gene coding for each protein sequence. All the states share the same gene and consequently part of their structure, the amino acid chain. They are functionally related, often even reversibly transformable into each other. A basic molecule entry captures this concept and is the class of all states of a protein. These states are different molecules, and we store them as different molecule entries. As molecules, they can be used in a pathway assembly. We store general information like the amino acid sequence in the basic molecule entry and link its states to it. In the simplest case there are only two states- an inactive one, and an active one. In other cases, there are more. For example, a transcription factor can be
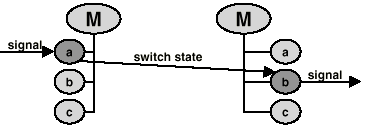
to name a few possibilities. The same protein will exhibit distinctly different signaling functions in these three states. For example in state 1 it will be susceptible to phosphorylation, in state 2 to translocation into the nucleus or dephosphorylation and in state 3 it will activate transcription. The number of states for a molecule is the product of the number of its modified forms and the number of locations where it is found. Only compounds which share the same location interact in nature. It is impractical to enter a separate state for each location. Most molecules can be found in several tissues, at several development stages, in several cellular compartments, several organs and several celltypes. To enter a state for each possible combination would lead to an explosion in the number of states, and redundancy in the reactions. This problem is circumvented by using a list of positive and negative locations that is linked to the basic molecule. In each state, the molecule is available only for a subset of all reactions for that molecule. Receiving a signal changes the molecule's state, usually leading to a new state from which reactions are triggered (Fig.2).
Motifs are structural and functional features of a protein and are often responsible for its signaling behavior. A single protein can have several signaling motifs. Motifs are listed in the feature field together with position information and references. More detailed information about the data model of TRANSPATH can be found at: "Consistent re-modeling of signaling pathways and its implementation in the TRANSPATH database" Genome Inf. Ser. 15, 244-254. [Pubmed] [.pdf]
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||