AliBaba2 is a program for predicting binding sites of transcription factor binding sites in an unknown DNA sequence. Therefore it uses the binding sites collected in TRANSFAC. AliBaba2 is currently the most specific tool for predicting sites.
What is it ?
![]()
AliBaba2 is a program for predicting binding sites of transcription
factor binding sites in an unknown DNA sequence. Therefore it uses the
binding sites collected in TRANSFAC. AliBaba2 is currently the most specific
tool for predicting sites.
Why is it ?
![]()
In predicting binding sites we face several problems as the binding
sites are short and can vary heavily. Mostly prediction is done by comparing
predefined matrices with an unknown sequence. But this leads to several
questions:
How does it work ?
![]()
The idea is NOT to use the predefined matrices available in databases
and in literature. Instead we start directly at the known binding sites.
Additionally we need a classification of these sites which is luckily also
available in TRANSFAC. AliBaba2 is a little process which consists
of three steps.
Parameters
![]()
You control the reliabiliy by using several parameters. Dont worry:
they are all already evaluated and the best parameter settings are determined
for the general case. Still you should briefly know what they do in case
you want.
Pairsim: for step 1 of the process, the pairwise alignment, we have to determine which sites we align to our unknown sequence. Pairsim is the threshold from which on we regard a known site as similar to out unknown sequence. Similarity is calculated like in the following example:
Factor class level: in step 2 of the process we have to tell AliBaba2 which sites belong to each other. Therefore the classification of transcription factor binding sites in TRANSFAC is used. This classification is a hierarchie consisting of 6 levels.
| Level | Name | Criterion | Example |
| 1 | Superclass | General topology of DBD | Zinc-coordinating |
| 2 | Class | Structural blueprint of DBD | Zinc finger nucl. recept. |
| 3 | Family | Functional criteria | T3R/RAR |
| 4 | Subfamily | Sequence similarity of DBD | RAR |
| 5 | Genus | According to factor gene | RAR-b |
| 6 | Factor "species" | Initiation/splice variants | RAR-b 1, RAR-b 2 |
In default setting level 4 is used to aggregate the binding sites in sets. This showed best results, but perhaps levels 3 or 5 are more interesting to you.
Matrix width: For step 3 of the process we have to determine the width of the matrix. Widths of 10 or 12 bp showed similar and optimal results. 12 bp was on the average a bit better. 10 bp is e.g. a bit better for SRF sites.
Matrix conservation: The quality of the matrices constructed in step 3 of the process is measured as the average information content, which is commonly named conservation. So a conserved matrix forms a very reliable prediction, while a low conserved matrix makes a more general statement. The more sites a matrix containts the lower the conservation is. AliBaba2 builds matrices till their conservation drops below the given parameter value. 70 % is a rather lazy restriction, 75 % is the default value and 80% showed to be rather strict.
Number of sites (support): The minimum number of sites of which a matrix is build is given here. The more sites a matrix contains the more reliable a prediction is. Generally it is believed the more sites you take the better results you get. But this is actually not true ! If you e.g. average all sites of SRF you will not get a good description of SRF, instead you get just noise. So analysis of sites has shown, that it is necessary to construct highly specific predictions for sites. Therefore it is recommended to keep the minimum number of sites, also called the support, low !
Similarity of seq to matrix: It is not only important to build matrices with a high conservation. Also the matrix must be similar to the sequence analysed. This similarity is measured using the Berg and von Hippel similarity. The similarity is measured in percent. 100 % means that the most often occuring nucleotides in matrix (the matrixs consensus) are the same like in the unknown sequence. 1% means that the unknown sequence is just similar to the matrix. Using the Berg and von Hippel measure has one great advantage: you can be sure that there never will be a nucleotide in the unknown sequence which contradicts a completely conserved nucleotide in the matrix. Thus we can be sure to get reliable predictions.
Performance
![]()
To evaluate the recognition performance
of AliBaba2, it has been compared to MatInspector public on TRANSFAC 3.5
educational as a freely available test set. MatInspector was used in both
settings "selected matrices" and "all matrices". The matter of reliability
can be measured as specificity and sensitity. For specificity the number
of predictions in 500 bp of exons is counted. For a count in sensitivity
a binding site contained in TRANSFAC has to be recognised without using
that one site (Jack-Knife test). Varying the threshold for MatInspector
and the conservation for AliBaba2 yields the graphs in Fig. 1. Result of
the comparison is that AliBaba2 clearly has a higher sensitivity as well
as a higher sensitivity / specificity ratio. In all specificity ranges
it constantly can detect about 500 binding sites more than MatInspector.
Fig. 2 shows the runtime of AliBaba2 when scanning a 30 kbp DNA sequence
gained from concatenating several promoter sequences on a 400 Mhz Pentium
II PC.
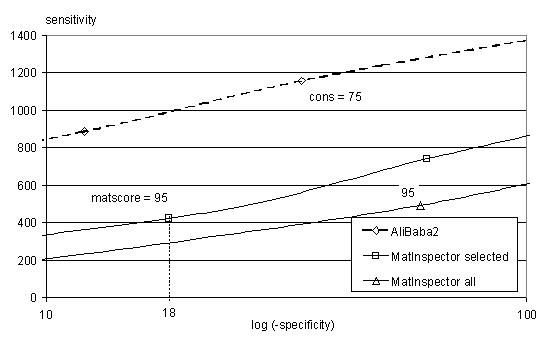
Fig.1: Comparing AliBaba2 to MatInspector
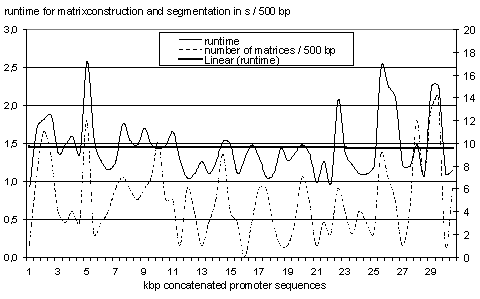
Fig.2: Runtime of AliBaba2 for 500 bp
Also see: Binding of OCT-1 from PDB