Project
application, 2006
HepatoSys - Kompetenznetz Systembiologie
des Hepatozyten
HepatoPath platform:
Building comprehensive regulatory network of hepatocytes through integration of
hormonal, gene regulatory, signal transduction and metabolic pathways.
Table
of contents
HepatoSys - Kompetenznetz
Systembiologie des Hepatozyten
1. Topic and aim of the project
Steps of the planned approach:
2. State of science and technology
and previous work of the consortia partners
Hormonal regulatory
network of liver cells
The innate
immune system in liver disease.
Analysis of
regulatory interactions and network structure
4. Novelty and attractiveness of
the product/method of resolution. Innovation aspects
5. Economic impact and market
potential
6. Applicant, contributing partners
and their expertise
Partner 2:
Department of Bioinformatics, University Göttingen (Wingender)
Partner 3:
Fraunhofer Institute of Toxicology and Experimental Medicine, Hannover, Germany
(Borlak)
Partner 5: Iinstitute of
Biochemistry, University of Cologne (Schomburg)
Partner 6:
Department of Gastroenterology & Endocrinology, University of Göttingen
(Ramadori)
Partner 7: Dept. Clinical and Experimental Endocrinology,
University of Goettingen (W. Wuttke)
Partner 8:
Department of Bioinformatics, University Freiburg (Backofen)
8. Detailed description of the work
plan and the contribution of each working group
WP1. Gene
expression analysis tools
WP2.
Signal transduction and transcription regulation network
WP3.
Metabolic and hormonal network_
WP4.
Network analysis and integration.
9. Detailed
costs projection (3 years)
Dissemination and standardization
activities
Abstract
Modeling the integral activity of huge
regulatory networks of multicellular organisms is a great challenge of modern
systems biology. Many advanced techniques have been employed in the last years
to model and simulate genetic regulatory systems, including: Boolean and Bayesian networks, their generalization
- Petri nets, differential equations, stochastic equations, the pi-calculus as
a formal language, and rule-based formalisms (reviewed in de Jong, 2002).
However, any simulation attempts hardly make any sense if the regulatory
network behind is not complete in their essential elements. To achieve the
ultimate goal of systems biology of building the virtual representation of the
cell and the whole organism in order to be able to perform the
"computational experiments" we must reconstruct the core regulatory
system in its maximal possible comprehensiveness. So, the goal of this project
is to build a most comprehensive regulatory network of the hepatocyte that will
include, first of all, gene regulation network, signal transduction and
metabolic as well as hormone and cytokine network of hepatocytes in various
developmental stages and physiological situations of normal and pathological
states of the organism. The regulatory
networks reconstructed in this project will provide the basis for building the
quantitative models and dynamic simulation in close cooperation with modeling
teams of the HepatoSys research framework.
Being the biggest
endocrine gland in our body, the liver is a control station of energy
homeostasis, metabolism/detoxification potential and the hormone system.
HepatoSys research framework puts its focus on modeling of two important
processes in liver: detoxification and regeneration. The ultimate goal is to
perform quantitative modeling of molecular-genetic mechanisms controlling these
processes in the liver cells to be able to understand their integral activity
in normal physiological situations as well as under metabolic pathophysiological
transdifferentiation and deregulated endocrine signaling processes
characteristic for various liver diseases.
As an integral
part of HepatoSys framework, the goal of our HepatoPath platform will be to
build the integrated regulatory networks of liver cells and populate it with
quantitative data using unique combination of modern experimental and
computational techniques. Starting from high throughput experimental
technologies of profiling the transcriptome, proteome, lipidome and metabolome,
we are going to contribute a novel approach to perform knowledge driven
reconstruction of integrity of hormonal, gene regulatory, signal transduction
and metabolic networks of hepatocytes. The core computational biology
technologies in our hands are the world leading databases: TRANSFAC on
transcription factors and their target sites, TRANSPATH on signal transduction
pathways (www.biobase.de), BRENDA on enzymes functional data and metabolic
reactions (www.brenda.uni-koeln.de) as well as novel databases: EndoNet (http://endonet.bioinf.med.uni-goettingen.de/)
on endocrine networks and miRNAdb on gene regulation via micro RNA (under
construction). The databases are accompanied by highly effective bioinformatics
tools for promoter analysis and network reconstruction. The project will be
also in close collaboration with international consortia in the field of
metabolomics such as the European Lipidomics Initiative (www.lipidomics.net).
First of all, in
the HepatoPath platform, we are going to extend the central intracellular
regulatory pathways important for hepatocyte regeneration and detoxification,
such as, JAK/STAT, SMAD, NF-kappaB,
Wnt/beta-catenin, AhR/ARNT, and
regulatory networks of cell cycle and apoptosis. Four extension frontiers
will be in our focus, that are absolutely essential for comprehensiveness of
pathways and the realistic chances for successful quantitative modeling: 1)
hormonal, cytokine and chemokine dependent regulation of the entry points of
the signaling pathways; 2) transcriptional regulation of the genes encoding the
components of the pathways and the mostly undiscovered multiple target genes
for the transcription factors involved - the source of often unconsidered
multiple feedback loops in the signaling pathways; 3) regulated metabolic
pathways leading to the catabolism and anabolism of hormones and energy; 4)
regulation through micro-RNAs.
Four main methods will be used by
the members of the consortia to achieve the goal of reconstructing the most
comprehensive regulatory network of hepatocytes: 1) modern high-throughput
techniques to measure the dynamics of transcriptome, proteome, lipidome and
metabolome; 2) expert annotation of the pathways through text mining and manual
curation of the vast body of scientific literature; 3) applying the most
advanced molecular biological techniques to detect protein-protein and
protein-DNA interaction in vivo, such as chromatin IP, and knowledge-driven
ChIP-on-chip approaches; 4) sophisticated computational methods, based on
machine learning techniques, to discover signals in gene regulatory regions and
algorithms of network analysis to find key nodes of the pathways.
Objectives.
1. To build the most
comprehensive regulatory network of the hepatocytes including gene regulation,
signal transduction and metabolic as well as hormone and cytokine network of
hepatocytes in the developmental stages and physiological situations of liver
detoxification response. Extension of the network towards liver-specific
pathological phenotypes such as steatosis/non-alcoholic steatohepatitis and
toxic liver injury and de-differentiation/regeneration.
2. To develop a novel
approach for modelling of complex biological systems by integration of
intracellular regulatory and metabolic pathways with the network of
communication between different populations of liver cells and soluble
components of the blood compartment in the context of the endocrine system of
the whole organism.
3. To build a new
knowledge-driven and data-rich platform that expands the logic of the current
HepatoSys research framework and provides network data to the consortia in
order to facilitate the quantitative modeling of hepatocytes.
Participants
P1 BIOBASE GmbH, Wolfenbüttel (Dr. A. Kel).
P2 UKG-G, University of
Göttingen, Dept. Bioinformatics, Göttingen (Prof. Dr. E. Wingender).
P3 ITEM, The Fraunhofer Institute of
Toxicology and Experimental Medicine, Dept. Drug Research and Medical
Biotechnology, Hannover (Prof. Dr. J. Borlak).
P4 UREG, Institute for Clinical Chemistry
and Laboratory Medicine University Hospital Regensburg, Regensburg (Prof. Dr.
G. Schmitz)
P5 IBUC, Institute of Biochemistry,
University of Cologne (Prof. Dr. D. Schomburg,)
P6 PUKG-G, University of Göttingen, Dept.
Gastroenterology and Endocrinology, Göttingen (Prof. Dr. G. Ramadori)
P7 UKG-E, University of Göttingen, Dept.
Clinical and Experimental Endocrinology, Göttingen (Prof. Dr. W. Wuttke).
P8 IIF, Institute of Informatics, University
of Freiburg, Freiburg (Prof. Dr. R. Backofen).
1. Topic and aim of the
project
Nowadays, we experience a shift in system
biology research from the study of single signal transduction pathways to
increasingly complex regulatory networks (Bornholdt, 2005). Detailed predictive
models of large regulatory networks could revolutionize our understanding of
complex biological systems like liver cell and would tremendously facilitate
the study of complex diseases, yet such models are not fissile to create. One
reason is that experimental data for large genetic systems are incomplete;
another is that large genetic systems require novel modeling approaches. Therefore, to bring the dream of system
biology closer to reality, the aim of the HepatoPath project is twofold.
First, to collect
all available data and generate missing experimental data on the
molecular genetic processes we are going to model. This includes,
characterizing the circuit wiring on all levels of regulations of cellular
processes: reception of extracellular hormone and cytokine signals provided by other
cells of the organism; transduction of the signals to the nucleus leading
to regulation of transcription, processing and translation of genes involved in
further regulatory processes and in regulation of metabolic pathways providing
cellular response to the signal and communication of the cell with other cells
of the organism. We are going to collect all this types of data for regulatory
pathways important for hepatocyte regeneration and detoxification, centered
around such important signal transduction molecules and transcription factors
as, JAK/STAT, SMAD, NF-kappaB, Wnt/betta-Catenin, AhR/ARNT, and on regulatory
networks of cell cycle and apoptosis. We will vastly extend the currently used
,rather limited, textbook knowledge on these pathways at the "top"
and at the "bottom": by adding hormonal regulation and transcription
regulation respectively. We will link these pathways to the network of the
metabolism of energy, toxins and hormones resulting in the most comprehensive
network of hepatocytes. We will also complement the study of intracellular
networks of hepatocytes with the analysis of circulating peripheral blood
monocytes as the hematopoietic percursor cells for Kupffer cells and the plasma
components. They will be used as correlating biological sources for liver
energy metabolism, detoxification of endogenous and foreign compounds and
immune mechanisms related to liver regeneration. We will generate the quantitative
and semi-quantitative data as far as possible in order to support maximally the
efforts on dynamic modeling of the networks. To do that will apply most modern
methods of experimental molecular biology and biochemistry for generation and
validation of the qualitative and quantitative network data. We will use a
combination of modern functional genomic techniques including gene expression
analysis, proteomics analysis, mediator metabolomics, analysis of chromatin
structure using significantly improved "ChIP-on-chip" method,
analysis of protein-protein interactions and most importantly innovative
knowledge-based bioinformatic techniques based on databases and methods of
artificial intelligence for computational identification of missing components
of the pathways followed up by experimental validation.
Second aim of HepatoPath
project is to extend existing and develop novel approaches suitable
for modeling of large regulatory networks. In the course of realization of this
project, powerful algorithms and tools will be developed to automate analysis
of data coming from DNA-microarrays, proteomics, metabolomics and lipidomics
experiments in order to extended the metabolic and signal transduction networks
and populate them with quantitative data. In the current project we will link
the intracellular networks with the hormonal, cytokine/chemokine and small
molecule mediator networks of the whole organism (Fig.1). By means of
topological graph analysis and information flow analysis of the network we will
identify the key controlling components and the key circuit motifs of the
system.
HepatoPath
platform will provide all this information on the reconstructed networks and
key node prediction to all members of HepatoSys consortia. The predictions will
be further validated by the modeling approaches developed in the HepatoSys
modeling platform. We will use selected endocrine disruptors on liver function
and apply RNAi in order to bring disturbances to the networks and to validate
the dynamic modeling results. Modelling of how the liver processes integrate
with all other organs of the organism by exchanging molecular signals is a
novel concept which will enhance the power of the already running HepatoSys
program in regard to the aims of Systems Biology
|
|
|
Fig. 1. Performing
knowledge-driven analyses of expression array data, we detect in-silico
those molecular changes that are not seen directly on the arrays. |
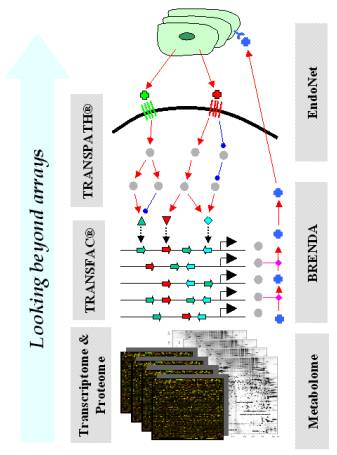
Steps
of the planned approach:
1. Build
comprehensive regulatory network of signal transduction integrated with
metabolic and hormonal and cytokine network by collecting all available data.
2. Analysis of
transcriptomics and proteomics data (using mircroarrays macroarrays, SAGE,
differential display and 2D display).
3. Prediction of
transcription factor binding sites and composite modules in the promoters of
gene regulatory clusters.
4. Prediction of key
nodes, by searching for common key regulators in the regulatory network
upstream of the differentially expressed genes and their transcription factors.
5. Experimental
validation of the TF-binding sites ChIP on chip and validation of key nodes by
RNAi.
2. State of science and
technology and previous work of the consortia partners
Integral activity of all processes in
hepotocytes are controlled by large regulatory network of tightly interlinked
gene regulatory, signal transduction, metabolic and hormonal pathways. A lot of
details on the structure of these networks have been elucidated in thousands of
experimental works in the previous years, still the are gaps in the knowledge
limiting modeling efforts of system biology.
Gene regulatory networks involved in liver differentiation,
regeneration, detoxification and lipid metabolism.
Gene expression is mainly regulated at the
transcriptional level through sequence-specific binding of transcription
factors (TFs) to their target sites in regulatory regions of genes, where the
combination of these sites and bound TFs and co-regulatory proteins as well as
the basal transcriptional machinery provide the required specificity.
TFs involved in liver-specific gene
regulation vary in terms of their structure and function. Some TFs are
liver-enriched, others are ubiquitous; some of TFs are activated in response to
extracellular stimuli; others are constitutive. Cooperative action of a great
number of TFs provides combinatorial transcriptional regulation of gene
expression in hepatocytes.
A simplified view
on transcription regulatory networks in liver differentiation, regeneration and detoxification is provided on Fig. 2.C/EBPs and HNFs are
master-regulators of hepatocyte
differentiation and regeneration.TFs of the AP-1 s are important regulators of
cell cycle and are closely involved in differentiation and regeneration as well
as detoxification. Tumor suppressor p53, via transcriptional control of a great
number of target genes regulates DNA repair, cell cycle checkpoints, apoptosis,
reversible and irreversible cell cycle arrests.
|
|
|
Fig. 2. C/EBP, HNF and the nuclear receptor gene
regulatory networks involved in the regulation of differentiation,
regeneration and detoxification in liver. |
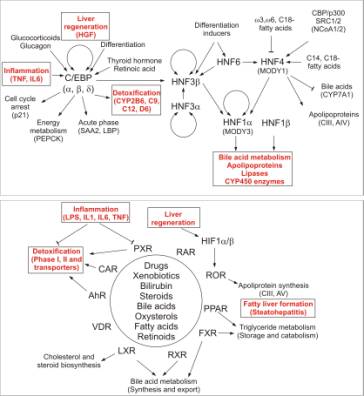
The best studied TFs in toxicology are
Ah-receptor (AHR), a member of helix-loop-helix-PAS domain TFs, and several
members of the nuclear receptor family including the pregnane-X-receptor (PXR),
constitutive androstane receptor (CAR), liver X receptor (LXR), farnesoid X receptor (FXR), PPAR, ROR. These factors
coordinately regulate gene batteries of cellular defence mechanisms and all
three phases of detoxification.AHR is known to function as a heterodimer with
Arnt; members of the nuclear receptor family form heterodimers with RXR.PPARs,
RXRs, the bile acid activated FXR, Arnt as well as sterol-regulatory element
binding proteins (SREBPs), are involved in the regulation of lipid metabolism.
Signal transduction pathways
Being the
dominant cell type in the liver, hepatocytes exist and function in the context
of other cells of the liver as well as cells of many other organs. In response
to various extracellular signaling molecules
such as hormones, cytokines, chemokines, a number of signal transduction
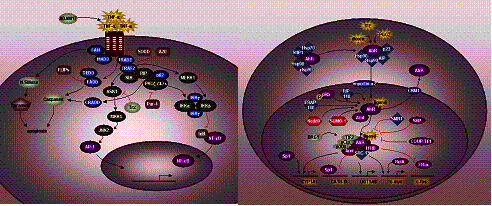
pathways are actived in hepatocytes. On the Fig. 3 we present principle schemas
of two signal transduction pathways important for liver regeneration and
detoxification taken from the TRANSPATH(R) database (Krull et al., 2006).
|
|
|
Fig. 3. a)
TNF-alpha pathway; b) AhR-signaling / Xenobiotic pathway |
Hormonal
regulatory network of liver cells
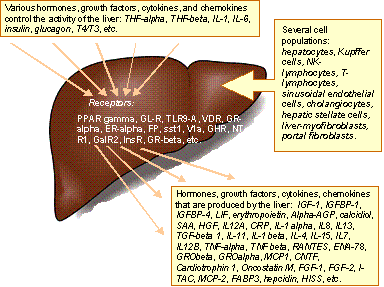
The liver is an important control
site of hormone homeostasis. Several hormones are degraded (e.g. insulin,
glucagon, thyroids and steroids) upon passage through the liver. Some hormones
are activated (e.g., conversion of T4 to T3 by hepatic deiodases), or
synthesized (e.g., insulin-like growth factors, IGFs, and several of their
binding proteins, IGFBPs) in the liver. The metabolic and regulatory pathways
serving for all these functions are subject to control by metabolite levels,
and circulating hormones. These different levels of control are strongly
interlinked in many ways. o model how the liver and hepatocytes processes
integrate with all other organs by exchanging molecular signals, we would like
to combine different experimental approaches with systematic collecting the
data in a proper database, i.e. EndoNet database (Potapov et al., 2006) on
intercellular regulatory pathways.
|
|
|
Fig. 4 Being the key player in maintenance of the metabolic balance,
detoxification, hormone homeostasis, exocrine and endocrine activities in a
human body, the liver is in the centre of intensive cell-to-cell and
tissue-to-tissue communications which are provided via numerous signaling
molecules (hormones, cytokines, chemokines, etc.) and their specific
receptors. |
The innate immune system in liver disease.
Liver contains multiple cell populations,
which are key components of the innate immune system: resident macrophage
populations termed Kupffer cells, NK cells, T-cells, and lymphocytes that
coexpress T- and NK- cell receptors. Hepatic Tcells produce IFN-gamma,
TNF-alpha, IL-2, and/or IL-4, but little or no IL-5, while NK-cells produce IFN-gamma
and/or TNF-alpha only. Kupffer cells are derived from circulating blood
monocytes that arise from bone marrow progenitors. Kupffer cells also generate
various immune defence products, including cytokines, prostanoids, nitric
oxide, and reactive oxygen intermediates. These molecules autoregulate the
phenotypic characteristics of Kupffer cells and also activate signal
transduction pathways in other liver cells.
Analysis of regulatory interactions and network
structure
New experimental high-throughput
approaches have been introduced to study the gene regulatory (and other
molecular) mechanisms of complex diseases. Among them are the ChIP (Chromatin
Immuno Precipitation)-on-chip method for the identification of in vivo
target genes for various TFs and RNAi approaches (gene silencing by small
double-stranded interfering RNAs) for the functional elucidation of selected
genes. For analysis of microarray data, various computational methods such as
hierachical clustering allow to allocate genes which are coregulated in time or
in response to specific treatments, into expression groupings called regulons.
Mapping of the clusters onto known metabolic, gene regulatory or signalling
pathways helps to reveal functional effects of the observed changes in gene expression.
In the work of P1, P2 and P5 over the many years the data
of the regulatory pathways are systematically collected in specialized
databases (TRANSFAC, TRANSPATH, TRANSCompel, BRENDA) that put the cornerstones in
this field (Matys et al., 2006; Krull et al., 2006; Schomburg et al., 2004).
Recently P2 came up with a new important database on intercellular
endocrine communication networks (EndoNet)
(http://endonet.bioinf.med.uni-goettingen.de/). (Potapov et al., 2006).
The results from
clustering analysis are limited since the correlation of gene expression does
not reveal "causality" in regulatory mechanisms. Posttranscriptional
and posttranslational regulation must be taken into account as well. Novel
methods of causal interpretation of gene expression data are necessary to draw
biological conclusions on the mechanisms and to select reasonable target genes.
This can be done by analysis of promoters and other regulatory regions of the
genes in co-expressed clusters. In the work of P1, P2 and P8
novel algorithms have been developed for analysis of promoter sequences and for
identification of TF binding sites and their specific compositions using
positional weigh matrix (PWM) and HMM approaches, various structural and
contextual features of promoter DNA and by applying sophisticated machine
learning techniques and genetic algorithm [Kel2005]. Since all site recognition
algorithms suffer from high prediction errors, additional evidence for the
predictions can be obtained by comparative analysis of genomic sequences,
phylogenetic footprint which is developed by P1 and P2
[Cheremushkin2003; Sauer2006]. Further improvement of these methods are
required by combining it with ChIP-on-chip data.
In order to model
dynamics of regulatory systems one partly requires information about binding
affinity of transcription factor to promoter sequences of particular genes. A
couple of computational approaches have been developed to model quantities of
binding affinities: [Udalova2002], that requires a dissimilarity measure for
known binding sequences, [Stormo2000], that uses PWMs as are good estimations
for binding affinity. P8 is working now to extend current solutions with
additional binding site features, such as local structural parameters, to
access the binding affinity.
Analysis of the
structure of large regulatory network using graph theoretical algorithms are
very promising for revealing key regulatory components of the network. Still
the problems arises due to the extremely high false positive rate of the
computationally reconstructed paths through the networks. P1 has
developed powerful algorithms of structural analysis of the network by
modelling of the chains of consecutive reactions using Markov models
[Kel-Margoulis In: Information Processing and Living Systems2005].
3. Contribution of the
project to the main focus of the program "Systeme des Lebens -
Systembiologie" of the German Government
This project contributes to the aims of
the program by providing the new platform HepatoPath. This platform will
significantly help to understand the biological modelsystem of hepatocytes. The
focus of this platform is to provide understanding of the regulatory network of
liver cells with respect to the integration of intracellular and intercellular
regulatory pathways. It should be pointed out that the extension to the
modeling of the liver cells in consideration of their tasks with other parts of
the organism (the intercellular pathways) brings a new quality and novel
concept into the HepatoSys program and fills a gap to the higher level of
systems biology. The program is thereby supplemented with a better
understanding of the central role and the functions this modeling system plays
in higher organisms. During this approach new concepts and methods are
developed that can be applied later to other modeling systems of systems
biology that will facilitate their understanding.
4. Novelty and
attractiveness of the product/method of resolution. Innovation aspects
A number of cutting edge methods and tools
is available in this consortium for the generation of a large amount of data on
of gene regulatory, signal transduction, metabolic and hormonal networks. The
challenge is now to bring these different methods together and to combine them
in a way that data generation and analysis becomes most cost- and
time-efficient. Our approach is based on combination of the most innovative
knowledge-based bioinformatic techniques based on databases and methods of
artificial intelligence and modern experimental methods of analysis of
chromatin structure and massive gene expression analysis. In the course of
realization of this project, powerful algorithms and tools will be developed to
automate analysis of data coming from microarrays and other high throughput
experiments. On the other side, experiments will be planned on the basis of
computational sequence analysis and modeling of the gene regulatory networks.
The intimate cooperation and quick feedback between experimental and in
silico approaches will allow us to direct and specify experiments according
to the hypothesis generated by bioinformatics studies, and finally to make them
more precise and less time-consuming. This is a clear step forward beyond the
nowadays practice of “hypothesis pure – data rich” studies.
Among the main innovations that we will
develop during this project is the significant improvement of the
“ChIP-on-chip” method by exploiting the advanced in silico site
recognition methods. This will allow us to construct genomic microarrays in a
very efficient way and to plan experiments on total investigation of genomic in
vivo targets for many different transcription factors. Our new method will
increase significantly the scope of these ChIP-on-chip experiments and make
them really high precision and genome-wide.
5. Economic impact and
market potential
The workflow proposed here comprises all
major steps in the early phase of drug development. Thus, if successful, the
whole package, including individual and integrated tools as well as the
corresponding services provided by the partners individually or in cooperation
with each other, will significantly speed up drug development in the
pharmaceutical industry. In the past, it was to be observed that many pure
bioinformatics approaches raised too high expectations about what could be done
for drug development and finally failed, frequently because of much too narrow
focussing on, e.g., modelling metabolic effects, rather than developing a
scientifically systemic view and a methodologically comprehensive approach as
we aim at with this proposal.
The approach can be easily applied to
other areas. Thus, all big pharmaceutical companies as well as smaller
companies involved in drug development form the potential market for the
envisaged offerings.
6. Applicant,
contributing partners and their expertise
Partner 1: BIOBASE GmbH (Kel)
Since its creation in 1997, BIOBASE's core
business is to maintain and distribute databases on gene expression regulation.
Its main product is TRANSFACâ, a database on
eukaryotic transcription factors (TF), their genomic binding sites and
DNA-binding profiles (PWMs). TRANSFACâ is accompanied by the sequence analysis tools
MATCH(tm) and PATCH(tm). TRANSCompelâ is a
collection of experimentally proven composite regulatory elements. TRANSPATHâ is a signal transduction pathways database. It comes
along with the tools PathwayBuilder(tm) and ArrayAnalyzer(tm).
PathwayBuilder(tm) provides the possibility to compose signaling paths,
pathways and networks out of the fragmented information stored in the databases
and to visualize them. ArrayAnalyzer(tm) is is a tool for the analysis of data
coming from microarray experiments. BIOBASE has proven expertise in the
prediction of binding sites for TFs, and also in the analysis of experimental
data. Researchers at BIOBASE have long standing experience in modeling of gene
networks involved in regulation of mammalian cell cycle. The model is enriched
by protein-DNA interactions that are predicted from the promoter analysis and
identification of TF binding sites. Modeling of such in-silico enriched networks provides the possibility to validate
the predicted regulatory interactions and to generate new hypotheses.
Dr. Alexander Kel is Senior
Vice-President Research and Development of BIOBASE. He has more than 20 years
research experience in Bioinformatics. He worked almost in all branches of
current Bioinformatics. He was involved in founding and developing several
databases on gene regulation and signal transduction (TRANSFAC, TRANSPATH, TRANSCompel,
TRRD), developing tools for analysis of gene regulatory regions and networks of
signal transduction. Currently, he is a scientific coordinator of INTAS project
funded by EU.
Selected
publications:
1. Kel A.E.,
Kel-Margoulis OV, Farnham PJ, Bartley SM, Wingender E. and Zhang MQ. (2001)
Computer-assisted Identification of Cell Cycle-related Genes: New Targets for
E2F Transcription Factors. J Mol Biol. 309: 99-120.
2. Kel A, Reymann S,
Matys V, Nettesheim P, Wingender E and Borlak J. (2004) A novel computational
approach for the prediction of networked transcription factors of Ah-receptor
regulated genes. Mol Pharmacol. 66: 1557-72.
3. Kel A, Konovalova
T, Valeev T, Cheremushkin E, Kel-Margoulis O and Wingender E. (2005) Composite
Module Analyst: A Fitness-Based Tool for Prediction of Transcription
Regulation. Lecture Notes in Informatics, Proceedings of the German Conference
on Bioinformatics 2005 (GCB 2005), Torda A, Kurtz S and Rarey M (eds.), Gesellschaft für Informatik, Bonn, 63-75.
4. Matys V,
Kel-Margoulis O, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I,
Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B,
Saxel H, Kel A and Wingender E. (2006) TRANSFAC(r) and its module
TRANSCompel(r): transcriptional gene regulation in eukaryotes. Nucleic Acids
Res. 34, D108-D110.
5. Krull M, Pistor S,
Voss N, Kel A, Reuter I, Kronenberg D, Michael H, Schwarzer K, Potapov A, Choi
C, Kel-Margoulis O and Wingender E. (2006) TRANSPATH(r): An Information
Resource for Storing and Visualizing Signaling Pathways and their Pathological
Aberrations. Nucleic Acids Res. 34, D546-D551.
Partner 2: Department of Bioinformatics, University
Göttingen (Wingender)
This group has long-standing experience in
network modeling and sequence analysis of promoter regions. Promoter models are
constructed that comprise defined cis-acting
sequence elements characterizing the promoter(s) of a certain group of genes.
Treating reported results of microarray analyses together with other available
information about the expressed genes, we search for distinguishing features of
the promoters of co-expressed genes. The application of such promoter models
enables to identify additional candidate genes belonging to the same regulatory
network. We investigated several defensive eukaryotic systems: (1)
antibacterial response of human lung epithelial cells to P. aeruginosa binding; (2) LPS-triggering (early response genes);
(3) MyD88-independent TLR4-triggered pathway; (4) MALP-2-triggered pathway. For
all considered systems the promoter models were provided. We developed several
new methods for promoter model construction and complemented them by novel
developments on phylogenetic footprinting approaches. In addition, the group has
accrued expertise in database development, in the integrative modeling of
different networks, and in establishing corresponding ontologies. Recently, a
database on intercellular communication networks (EndoNet) has been made
available by the group (http://endonet.bioinf.med.uni-goettingen.de/). EndoNet focuses on the endocrine
cell-to-cell signaling and enables the analysis of human intercellular
regulatory pathways. It aims at bridging the existing gap between known
genotypes and their molecular and clinical phenotypes, thus allowing
utilization of EndoNet in medical research.
Prof. Dr. Edgar
Wingender has coordinated and contributed to several national and international
bioinformatics projects since the start of bioinformatics funding by BMFT/BMBF
in 1993. Having established and headed the Research Group Bioinformatics at GBF
until 2002, he then accepted the call to the University of Göttingen where he
is now heading the Department of Bioinformatics at the Medical School. He is
founder and Scientific Director of BIOBASE GmbH. His expertise is in
bioinformatics of gene regulation and signal transduction.
Selected
publications:
1. Shelest E, Kel A
E, Goessling E and Wingender E. (2003) Prediction of potential C/EBP/NF-kappaB
composite elements using matrix-based search methods. In Silico Biol. 3: 71-79.
2. Potapov AP, Voss
N, Sasse N and Wingender E. (2005) Topology of mammalian transcription
networks. Genome Inf Ser. 16, 270-278.
3. Shelest E and
Wingender E. (2005) Construction of predictive promoter models on the example
of antibacterial response of human epithelial cells. Theor. Biol. Med. Model.
2, 2.
4. Chen X, Wu JM,
Hornischer K, Kel A and Wingender E. (2006) TiProD: The Tissue-specific
Promoter Database. Nucleic Acids Res. 34, D104-D107.
5. Potapov A, Liebich
I, Dönitz J, Schwarzer K, Sasse N, Schoeps T, Crass T and Wingender E. (2006)
EndoNet: An information resource about endocrine networks. Nucleic Acids Res.
34, D540-D545.
6. Sauer T, Shelest E
and Wingender E (2006) Evaluating phylogenetic footprinting for human-rodent
comparisons. Bioinformatics, in press.
"Übersicht über
bewilligte Drittmittelprojekte der Jahre 2002-2005":
The department participates in two German
government-financed projects: Helmholz Open Bioinformatics Technology (HOBIT,
since 2003) as well as the National Genome Research Network (NGFN2, since
2005). It takes part in two projects finaced by the EU: COMBIO (since 2004) and
TEMBLOR (2002 – 2005).
Partner 3: Fraunhofer Institute of Toxicology and Experimental Medicine,
Hannover, Germany (Borlak)
The Center of Drug Research and Medical
Biotechnology was founded in 1998 and now consists of 9 postdoctoral fellows (3
men and 6 women), 3 doctoral fellows (3 men) and 12 technicians (2 men and 10
women). In this Center a wide range of in vivo and in vitro systems are
available which enable problem solving and a mechanistic approach in
pharmacology, toxicology and cancer. Alongside routine tests in molecular
dosimetry, biochemical toxicology, xenobiochemistry, and some specialized
studies in endocrine toxicology, gene expression patterns are also investigated
in pathology and pharmacological and toxicological assessments using advanced
molecular biology techniques.
We have gained in depth diligence in the
fields of genomics, proteomics, metabonomics, cell culture, ChIP-on-chip
technology and bioinformatics (in silico analyses of gene expression data). We
collaborate with different European clinical centers and are able to get hold
of different patient tissues.
Selected
publications:
1. ·Borlak J, Meier
T, Halter R, Spanel R, Spanel-Borowski K. (2005) Epidermal growth
factor-induced hepatocellular carcinoma: gene expression profiles in precursor
lesions, early stage and solitary tumours. Oncogene. 2005 Mar
10;24(11):1809-19.
2. ·Niehof M, Borlak
J. (2005) RSK4 and PAK5 are novel candidate genes in diabetic rat kidney and
brain. Mol
Pharmacol.2005 Mar;67(3):604-11.
3. ·Kel, A., Reymann,
S., Matys, V., Nettesheim, P., Wingender, E. and Borlak, J. (2004) A novel
computational approach for the prediction of networked transcription factors of
Ah-receptor regulated genes. Mol Pharmacol. 2004 Dec;66(6):1557-72
4. ·Schrem H,
Klempnauer J, Borlak J. (2004) Liver-enriched transcription factors in liver
function and development. Part II: the C/EBPs and D site-binding protein in
cell cycle control, carcinogenesis, circadian gene regulation, liver
regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev.
56:291-330.
5. ·Thum T, Borlak J.
(2004) Mechanistic role of cytochrome P450 monooxygenases in oxidized low-density
lipoprotein-induced vascular injury: therapy through LOX-1 receptor antagonism?
Circ Res. Jan 9;94(1):e1-13.
6. ·Rütters, H.,
Zürbig, P., Halter, R. and Borlak, J. (2006) Towards a lung adenocarcinoma
proteome map – studies with SP-C/c-raf transgenic miceProteomics, accepted for
publication
Partner 4: Institute of Clinical Chemistry and Laboratory Medicine,
University of Regensburg (Schmitz)
The research of the Institute of Clinical
Chemistry and Laboratory Medicine is directed towards the molecular analysis of
metabolic diseases with focus on lipid homeostasis, organ transdifferentiation,
cellular detoxification and the central role of monocytes/macrophages in the
pathogenesis of high triglyceride/ low HDL syndromes. Our strategy combines
basic research with clinical studies enabling a rapid transfer of newly
identified gene candidates and biomarkers from basic science to clinical
diagnostics. The clinical part is related to the Regensburg Diabetes Endpoint
Prediction and Prevention Study (REDEPPS), an interdisciplinary project that
enables the establishment of large sample and data banks from patients with
different endpoints or co-morbidities of type 2 diabetes including liver
diseases such as steatohepatitis under GLP conditions. REDEPPS is embedded in
the Danubian Biobank Consortium (SSA DanuBiobank) that has been initiated by
partner 4. Both initiatives are members of the global P3G consortium for
standardization of biobanking. The group has a special interest and expertise
in transcriptional regulatory networks and bioinformatics of lipid homeostasis
and complex diseases in close collaboration with the newly founded Institute of
Functional Genomics (Prof. Oefner) and the Bavarian Genome Project (BayGene).
The available technologies include high throughput genotyping, DNA-microarrays
and Taqman PCR for genetic analysis and MALDI-TOF and the multicolour 2D-Gel
Typhoon system for proteomics. The center has established an elaborated
lipidomics platform with high performance gel-filtration chromatography
methods, tandem mass spectrometry (ESI-MS/MS, GC-MS), capillary
isotachophoresis and gradient gel analysis for lipoprotein subspecies and lipid
analysis. The institute is cofounder and partner of the European Lipidomics
Initiative (SSA ELIfe).
Sophisticated cellular life imaging
techniques (Discovery 1 high content screening system, Leica fluorescence life
time imaging and confocal microscopy) have been established and the group has
developed a series of multiparameter flow cytometric assays including flow-FRET
to study innate immunity receptors clusters on monocytes. In this context the
group has identified the ATP binding cassette transporter ABCA1 as a major
regulator of raft-microdomain dynamics and HDL metabolism in macrophages,
hepatocytes and enterocytes. Work from the SFB/Transregio 13 (Membrane
microdomains in disease) has lead to the discrimination of
sphingomyelin/cholesterol rich from cermaide/cholesteril rich raft microdomains in monocytes of patients
with lipid disorders, and lipidomic characterization of plasma from sepsis
patients revealed ceramide and lysophosphatidylcholine as novel sepsis
mortality parameters. Furthermore, the group has extensively studied epithelial
cell homeostasis in inflammatory bowel disease (IBD) and has identified a loss
of detoxification potential and reduced expression levels of the master
detoxification transcription factor PXR (pregnane-X-receptor) in the colon of
IBD patients. Mediator lipidomics and metabolomics techniques for
quantification of transcription factor ligands including nuclear receptor
cofactors such as bile acids, oxysterols and fatty acid derivatives have been
successfully developed over the last years.
Prof. Dr. med.
Gerd Schmitz, is the director of the Institute of Clinical Chemistry and Laboratory
Medicine and together with Prof. Dr. med R. Andreesen runs the local stem cell
center and immunehematological diagnostic center of the university hospital.
His research is directed towards the molecular analysis of metabolic and aging
disorders with focus on lipid homeostasis and the central role of epithelial
cells and blood cells in the pathogenesis of complex diseases. Our strategy
combines basic research with clinical research enabling a rapid transfer of
newly identified gene candidates and biomarkers from basic science to clinical
diagnostics. We are founding member of the Regensburg Diabetes Endpoint
Prediction and Prevention Study. The group has expertise in transcriptional
regulatory networks and bioinformatics of lipid homeostasis, transdifferentiation
and detoxification in complex diseases. The Institute has established a state
of the art transcriptomics and lipidomics platform and is cofounder of the
European Lipidomics Initiative (www.lipidomics.net).
Selected
publications:
1. Bodzioch M, Orso
E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S,
Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G,
Aslanidis C, Lackner KJ, and Schmitz G. (1999) The gene encoding ATP-binding
cassette transporter 1 is mutated in Tangier disease. Nat Genet. 22: 347-51.
2. Langmann T,
Liebisch G, Moehle C, Schifferer R, Dayoub R, Heiduczek S, Grandl M, Dada A,
Schmitz G. Gene expression profiling identifies retinoids as potent inducers of
macrophage lipid efflux. Biochim Biophys Acta. 2005 May 30;1740(2):155-61.
3. Liebisch G,
Drobnik W, Lieser B, Schmitz G. High-throughput quantification of
lysophosphatidylcholine by electrospray ionisation tandem mass spectrometry.
Clin Chem 2002;48: 2217-24.
4. Langmann T,
Mauerer R, Zahn A, Moehle C, Probst M, Stremmel W, Schmitz G. Loss of
detoxification in inflammatory bowel disease: dysregulation of pregnane X
receptor target genes. Gastroenterology 2004;49:230-8.
5. Orsò E, Broccardo
C, Kaminski WE, Bottcher A, Liebisch G, Drobnik W, Gotz A, Chambenoit O,
Diederich W, Langmann T, Spruss T, Luciani MF, Rothe G, Lackner KJ, Chimini G,
Schmitz G. Transport of lipids from golgi to plasma membrane is defective in
tangier disease patients and Abc1-deficient mice. Nat Genet. 2000;24:192
6. Drobnik W, Liebisch
G, Audebert FX, Frohlich D, Gluck T, Vogel P, Rothe G, Schmitz G. Plasma
ceramide and lysophosphatidylcholine inversely correlate with mortality in
sepsis patients. J Lipid Res. 2003 Apr;44(4):754-61.
"Übersicht über
bewilligte Drittmittelprojekte der Jahre 2002-2005":
|
Thema |
Name(n) |
Zuwendungsgeber,
Aktenzeichen |
Bewilligungs-zeitraum |
Mittel |
|
Quantifizierung
von bioaktiven Lipiden des Sphingo- und Glycerophospholipidstoffwechsels
mittels tandem-Massenspektrometrie |
G.
Liebisch |
DFG:
LI 923/2-1 |
ab Jan
2004 für 2(+1) Jahre |
BAT
IIa/2, 20.000,- € (+10.000 €) Sachmittel |
|
Konjugierte
Linolsäuren und verzweigtkettige Fettsäuren als Lipidantagonisten der
genexpression und therapeutische Targets in Darmepithelzellen und Monozyten |
G.
Schmitz |
DFG:
SCHM 654/9-2 (im Verbundprojekt Lipide) |
ab April
2005 für 2(+1) Jahre |
BAT IIa, BAT IIa/2, 37.000 € (18.500 €) |
|
Charakterisierung
von ZNF202 im Lipidmetabolismus |
S.
Heimerl, G. Schmitz |
DFG:
HE 4727/1-1 |
ab Aug
2005 für 2 Jahre |
BAT Va/b,
32.100 € Sachmittel |
|
Charakterisierung
der Expression, morphologischen Verteilung und Regulation von
ATP-binding-cassette Transportern in der Darmmukosa von Patienten mit
entzündlichen Darmerkankungen |
T.
Langmann, G. Schmitz |
DFG: SFB
585/A1 (Regulation von Immunfunktionen im Verdauungstrakt) |
01.01.2002
- 30.06.2005 |
BATIIa/2,
BATVb, 40.000 DM Sachmittel pro Jahr |
|
Die
Bedeutung von „Lamellar Bodies“ bei Störungen der intestinalen
Membranintegrität im Rahmen chronisch-entzündlicher Darmerkrankungen |
E.
Orso, G. Schmitz |
DFG:
SFB 585/A4 (Regulation von Immunfunktionen im Verdauungstrakt) |
01.01.2002
- 31.12.2005 |
BATIIa/2,
BATVb, 20.000 DM Sachmittel pro Jahr |
|
DNA/RNA
Analytik und Arraytechnologie |
G.
Schmitz |
DFG:
SFB 585/Z2 (Regulation von Immunfunktionen im Verdauungstrakt) |
01.01.2002
- 31.12.2005 |
BATIIa,
25.000 DM Sachmittel pro Jahr |
|
Analysis of ABCA1 interactive proteins
and raft domain association depending on genetic factors and pre-beta-HDL
composition |
G. Schmitz |
DFG: SFB TR13/A3 (Membrane Microdomains
and Their Role in Human Disease) |
01.01.2004
- 31.12.2007 |
2
BATIIa/2, 12.800 € pro Jahr |
|
Bedeutung
von humanen Monozyten/Makrophagen für die Immunpathogenese fakultativ
intrazellulärer Erreger am Modell von Francisella tularensis |
G.Schmitz |
Bundesministerium
für Verteidigung |
01.08.04 - 31.07.06 |
BATVb, 130.000 Sachkosten |
|
Central Facility for the Production of Stabilised Cellular Reference Standards
and External Quality Assessment in Clinical Flow Cytometry (EuroStandards) |
G. Rothe |
EU: QLRI-CT-2000-00436 |
01.09.00 - 28.02.04 |
BATIIa, 12.000 € Reisen, 39.000 € consumables |
|
Dietary Lipids as Risk Factors in Development
(DLARFID) |
G.
Schmitz, T. Langmann |
EU: QLK1-CT-2001-00183 |
01.01.02 - 31.12.04 |
BATIIa, BAT Vb, 10.000 € travel, 60.000 € consumables |
|
The European Lipidomics Initiative;
Shaping the life sciences (ELife) |
G.
Schmitz |
EU:
SSA 013032 |
01.01.05 - 31.12.06 |
|
Partner 5:
Iinstitute of Biochemistry, University of Cologne (Schomburg)
The University of Cologne is one of the
premier German institutions in the fields of protein bio-chemistry and
genetics. In Prof. Schomburg's group at the Institut für Biochemie research is
performed in the areas of enzymology, structural biochemistry and
bioinformatics. The Iinstitute of Biochemistry is also responsible for the development, maintenance
and curation of the BRENDA enzyme function database. The enzyme information
system BRENDA (www.brenda.uni-koeln.de), started in 1987, is the world's most
comprehensive enzyme function and property database and is made available to
the scientific community via a complex query system on the Internet and is
cu-rated with close links to the user community. The BRENDA site registers more
than 2 million hits per month and is queried by ca. 1000 different scientists
per day.
The group is also actively involved in
research on enzyme function. The contribution of the University of Cologne will rely on its
long-standing experience in development and maintenance of BRENDA as well as on
its active involvement in standardisation of biochemical terminology and
promotion of recommended scientific nomenclature.
Selected
publications:
1. Hofmann, O. &
Schomburg, D. (2005), 'Concept-based annotation of enzyme classes.',
Bioinformatics 21(9), 2059--2066.
2. Rahman, S.A.;
Advani, P.; Schunk, R.; Schrader, R. & Schomburg, D. (2005), 'Metabolic
pathway analysis web service (Pathway Hunter Tool at CUBIC).', Bioinformatics
21(7), 1189--1193.
3. Fleischmann, A.;
Darsow, M.; Degtyarenko, K.; Fleischmann, W.; Boyce, S.; Axelsen, K.B.;
Bai-roch, A.; Schomburg, D.; Tipton, K.F. & Apweiler, R. (2004), 'IntEnz,
the integrated relational en-zyme database.', Nucleic Acids Res 32(Database
issue), D434--D437.
4. Heuser, P.;
Wohlfahrt, G. & Schomburg, D. (2004), 'Efficient methods for filtering and
ranking fragments for the prediction of structurally variable regions in
proteins.', Proteins 54(3), 583--595.
5. Schomburg, I.;
Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G. & Schomburg, D.
(2004), 'BRENDA, the enzyme database: updates and major new developments.',
Nucleic Acids Res 32(Database issue), D431--D433.
6. Ehrentreich, F.
& Schomburg, D. (2003), 'Dynamic generation and qualitative analysis of
metabolic pathways by a joint database/graph theoretical approach.', Funct
Integr Genomics 3(4), 189--196.
"Übersicht über
bewilligte Drittmittelprojekte der Jahre 2002-2005":
Prof. Schomburg obtained a grant for the
establishment of the "Cologne University Bioinformatics Centre
(CUBIC)", an "Exploratory Project" grant in the framework of
NGFN2 on metabolomics/systems biology, an EU grant on the establishment of an
integrated enzyme database, is a member of two EU
networks of excellence, obtained a GIF
grant on protein structure determination, is a member of a Max-Planck research
school, obtained a grant from the Helmholtz-strategy fund as well as
DFG-grants.
Partner 6: Department of Gastroenterology & Endocrinology,
University of Göttingen (Ramadori)
The Dept. of Gastroenterology &
Endocrinology has a long tradition in basic liver cell research. The group has
a broad experience in the isolation and purification of the different liver
cell populations including hepatocytes, sinusoidal endothelial cells, hepatic
stellate cells, Kupffer cells, and inflammatory mononuclear cells. A series of
studies has established an important role of the non-parenchymal cells in
disease processes of the liver, particularly in pathomechanisms of toxic liver
injury and development of liver fibrosis. These works were funded by DFG, SFB
402. Currently, the research is
directed towards gene expression analysis in different developmental stages of
the liver and in different animal models of
liver diseases with a focus on liver regeneration. To this aim, several
animal models have been established at the Dept. including acute and chronic
liver injury (rat CCl4-model), gamma-irradiation of the rat liver, liver
regeneration after partial hepatectomy in rats and mice (incl. sham
operations), acute phase reaction (turpentine-oil rat model), liver
regeneration via hepatic stem cells (rat oval cell model by the modified
Solt-Farber protocol, AAF administration and partial hepatectomy). Hepatic gene
expression was studied in these models by a wide variety of analysis tools,
including SAGE analysis, micro- and macroarray analyses and differential
display. Resulting data were confirmed on a single gene basis by Real-time PCR
and Northern Blot analysis. Analyzing and comparing hepatic gene expression
under different in vivo conditions provides us the unique possibility to
characterize distinct genetic pathways involved in health, liver injury and/or
regeneration.
Prof. Dr. G.
Ramadori worked in internal medicine at the Free University Berlin and at the
University of Mainz with main scientific focus on basic liver research. He runs
the department of Gastroenterology at the University of Göttingen since 1992
and he is one of the initiators of the Sonderforschungsbereich 402 (SFB 402,
Collaborative Research Center 402) in 1993, and serves as the director of the
SFB since 2002. He is also co-founder of the Graduiertenkolleg 335 (GRK 335) at
the University of Göttingen. He is member of the Editorial Board of Laboratory
Investigation and of BMC-Hepatology, and he is an appointed reviewer for the
Deutsche Forschungsgemeinschaft (DFG), NIH, Wellcome Trust, MRC, SNRC (Swiss
National Research Council), Austrian Council for Research and Technology
Development .
Selected
publications:
1. Armbrust T,
Kreissig M, Tron K, Ramadori G (2004) Modulation of fibronectin gene expression
in inflammatory mononuclear phagocytes of rat liver after acute liver injury. J
Hepatol 40:638-645
2. Batusic DS, Armbrust
T, Saile B, Ramadori G (2004) Induction of Mx-2 in rat liver by toxic injury. J
Hepatol 40:446-453
3. Batusic DS, Cimica
V, Chen Y, Tron K, Hollemann T, Pieler T, Ramadori G (2005) Identification of
genes specific to "oval cells" in the rat 2-acetylaminofluorene/partial
hepatectomy model. Histochem Cell Biol 124:245-260
4. Christiansen H,
Batusic DS, Saile B, Hermann RM, Dudas J, Rave-Frank M, Hess CF, Schmidberger
H, Ramadori G (2005) Identification of genes early responsive to
gamma-irradiation in rat hepatocytes by cDNA array gene expression analysis.
Radiation Research
4.
5. Christiansen H,
Saile B, Neubauer-Saile K, Tippelt S, Rave-Frank M, Hermann RM, Dudas J, Hess
CF, Schmidberger H, Ramadori G (2004) Irradiation leads to susceptibility of
hepatocytes to TNF-alpha mediated apoptosis. Radiother Oncol 72:291-296
6. Cimica V, Batusic
D, Hollemann T, Chen Y, Pieler T, Ramadori G (2004) Transcriptome Analysis of
Early Stage of Rat Liver Regeneration In the Model of Oval Hepatic Stem Cells.
Biochem Biophys Res Commun
7. Cimica V, Batusic
D, Chen Y, Hollemann T, Pieler T, Ramadori G (2005) Transcriptome analysis of
rat liver regeneration in a model of oval hepatic stem cells. Genomics
86:352-364
8. Dudas J, Papoutsi
M, Hecht M, Elmaouhoub A, Saile B, Christ B, Tomarev SI, von Kaisenberg CS,
Schweigerer L, Ramadori G, Wilting J (2004) The homeobox transcription factor
Prox1 is highly conserved in embryonic hepatoblasts and in adult and
transformed hepatocytes, but is absent from bile duct epithelium. Anat Embryol
(Berl) 208:359-366
9. Haralanova-Ilieva
B, Ramadori G, Armbrust T (2005) Expression of osteoactivin in rat and human
liver and isolated rat liver cells. J Hepatol 42:565-572
10. Mihm S, Frese M,
Meier V, Wietzke-Braun P, Scharf JG, Bartenschlager R, Ramadori G (2004) Interferon
type I gene expression in chronic hepatitis C. Lab Invest 84:1148-1159
11. Novosyadlyy R,
Tron K, Dudas J, Ramadori G, Scharf JG (2004) Expression and regulation of the
insulin-like growth factor axis components in rat liver myofibroblasts. J Cell
Physiol 199:388-398
12. Ramadori G, Saile
B (2004a) Inflammation, damage repair, immune cells, and liver fibrosis:
specific or nonspecific, this is the question. Gastroenterology 127:997-1000
13. Ramadori G, Saile
B (2004b) Portal tract fibrogenesis in the liver. Lab Invest 84:153-159
14. Ramadori G, Saile
B: Hepatocytes. Hepatology, 2005, pp 1-31
15. Saile B, Eisenbach
C, Dudas J, El-Armouche H, Ramadori G (2004a) Interferon-gamma acts
proapoptotic on hepatic stellate cells (HSC) and abrogates the antiapoptotic
effect of interferon-alpha by an HSP70-dependant pathway. Eur J Cell Biol
83:469-476
16. Saile B, DiRocco
P, Dudas J, El-Armouche H, Sebb H, Eisenbach C, Neubauer K, Ramadori G (2004b)
IGF-I induces DNA synthesis and apoptosis in rat liver hepatic stellate cells
(HSC) but DNA synthesis and proliferation in rat liver myofibroblasts (rMF).
Lab Invest 84:1037-1049
17. Tron K,
Novosyadlyy R, Dudas J, Samoylenko A, Kietzmann T, Ramadori G (2005)
Upregulation of heme oxygenase-1 gene by turpentine oil-induced localized
inflammation: involvement of interleukin-6. Lab
Invest 85:376-387
"Übersicht über bewilligte Drittmittelprojekte der Jahre 2002-2005"
- Sonderforschungsbereich 402 (SFB 402), Molekukare und zelluläre
Hepatogastroenterologie, 3. Förderperiode ab 1.1.2004 -31.12.2006
Projekt:
„Reparaturprozesse in der geschädigten
Leber - Einfluss von Zellinteraktionen auf die Überlebensmechanismen von
aktivierten hepatischen Sternzellen und Leber Myofibroblasten“ Ramadori, Saile
Projekt: „Entwicklung und
Regeneration der Leber: Molekulare Mechanismen bei Proliferation und
Differenzierung von Hepatozyten im Verlauf der embryonalen Lebereintwicklung
und bei der Regeneration der adulten Leber in Vertebraten“
Pieler, Ramadori
- Graduiertenkolleg
335 (GRK 335)
“Molecular,
Cellular, and Clinical Biology of Internal Organs”
Projekt: “Molecular mechanisms of the cytokine-dependent
induction of the heme oxygenase-1 gene: In vivo and in vitro studies”
Projekt: “Role of hepatic IGF-I in liver fibrogenesis.”
- HepNet (Kompetenznetz Hepatitis, 2. Förderperiode,
ab 1.1.2005)
Projekt:
„Prognostische Marker und Mechanismen der
Fibrogenese und Strategien für deren Inhibition“ Ramadori
- Deutsche Krebshilfe
Projekt
„Molekulare Mechanismen der akuten und
chronischen strahlen-induzierten Leberschädigung“, Christiansen,
Schmidberger, Saile, Ramadori; ab 1.01.2006
Partner 7: Dept.
Clinical and Experimental Endocrinology,
University of Goettingen (W. Wuttke)
Experience in the co-ordination of EU- and
German Research Society (DFG) funded projects, referee for numerous national
(DFG, ministry of science) and international (NIH, GIF, NSF, MRE) funding
agencies.
More than 30 years of experience in
endocrine research with animal models and primary and permanent cell culture
systems suitable to study genomic and non-genomic actions of steroids and their
naturally occurring or synthetic analogues in steroid receptive organs
including the liver.
Methods to assess liver function at
transcriptional and functional levels are established in the laboratory (e. g.
DNA-microarrays and Taqman realtime RT-PCR , immunohistochemistry, RIA, ELISA)
Selected
publications:
1. Seidlova-Wuttke D,
Christoffel J, Rimoldi G, Jarry H, Wuttke W. Comparison of effects of estradiol
with those of octylmethoxycinnamate and 4-methylbenzylidene camphor on fat
tissue, lipids and pituitary hormones. Toxicol Appl Pharmacol. 2005 Dec 17; [Epub
ahead of print]
2. Supornsilchai V,
Svechnikov K, Seidlova-Wuttke D, Wuttke W, Soder O. Phytoestrogen resveratrol
suppresses steroidogenesis by rat adrenocortical cells by inhibiting cytochrome
P450 c21-hydroxylase.Horm Res. 2005; 64:280-286
3. Klammer H,
Schlecht C, Wuttke W, Jarry H. Multi-organic risk assessment of estrogenic
properties of octyl-methoxycinnamate in vivo A 5-day sub-acute pharmacodynamic
study with ovariectomized rats.Toxicology. 2005;215: 90-96.
4. Seidlova-Wuttke D,
Jarry H, Christoffel J, Rimoldi G, Wuttke W. Effects of bisphenol-A (BPA),
dibutylphtalate (DBP), benzophenone-2 (BP2), procymidone (Proc), and linurone
(Lin) on fat tissue, a variety of hormones and metabolic parameters: a 3 months
comparison with effects of estradiol (E2) in ovariectomized (ovx)
rats.Toxicology. 2005; 213: 13-24.
5. Schlecht C,
Klammer H, Jarry H, Wuttke W. Effects of estradiol, benzophenone-2 and
benzophenone-3 on the expression pattern of the estrogen receptors (ER) alpha
and beta, the estrogen receptor-related receptor 1 (ERR1) and the aryl
hydrocarbon receptor (AhR) in adult ovariectomized rats. Toxicology. 2004;
205:123-130.
Partner 8: Department of Bioinformatics, University Freiburg (Backofen)
The group has generally experience with
developing and applying bioinformatics methods for the detection of regulatory
sequences. A particularly relevant work of this group is the stochastic
modeling of transcription factor binding sites. We have developed a flexible
modeling framework which is based on Bayesian networks and thus able to
integrate various biological features (like chromatin structure) of binding
sites and interdependencies among these features. The distinguishing features
are retrieved using feature subset selection (FSS) algorithms which take a set
of TRANSFAC binding sites as input. The resulting binding site models have
shown to have a better predicting performance compared to common positional
weight matrices. We are currently developing a stochastic reasoning approach
which combines the prediction results of a set of single binding site models
with an expectation to find such hits. This expectation is based on evaluating
arbitrary biological data such as tissue information or neighboring binding
sites for co-acting factors, thus favoring binding site clusters. The
underlying reasoning machine is a specially designed Bayesian network which
possesses properties of Boolean networks.
Beside the transcriptional regulation,
another extensively researched topic of the group is the development of
(non-EST-based) approaches for detecting alternative splice forms. Alternative
splicing is one important way of post-transcriptional regulation. Within the
SFB 604 “Multifunctional Signaling Proteins”, we are currently investigating
how alternative splicing works as an modulator for signal transduction. The
group is using its modeling expertise to determine signals which favor
alternative splicing events, such as RNA sequence/structure motifs. Sequence
and RNA structure information together is used to discover novel motifs from a
set of input sequences (e. g. SELEX data).
Prof. Dr. Rolf
Backofen, established and headed the Chair in Bioinformatics at the University of
Jena, where he is participating in several projects in the Jena Centre of
Bioinformatics. He is also part of the SFB "Multifunctional Signalling
Proteins", the EU-network of excellence "REWERSE", and EU-STREP
project "EMBIO". In 2005, the accepted the offer for a chair in
bioinformatics from the University of Freiburg. His expertise is in algorithmic
bioinformatics.
Publications
2002-2005
1. Backofen R. and Will S. (2006) A constraint-based approach to fast and exact
structure prediction in three-dimensional protein models. Journal of Constraints, 11(1) To
appear.
2. Backofen R. and Siebert S. (2005) Fast detection of common sequence structure patterns
in RNAs. Journal of
Discrete Algorithms, To appear.
3.
Hiller M., Huse K., Platzer M., and Backofen R.
(2005) Creation and disruption of protein
features by alternative splicing - a novel mechanism to modulate function. Genome
Biol,
6(7):R58.
4. Busch
A., Will S., and Backofen R. (2005) SECISDesign:
a server to design SECIS-elements within the coding sequence. Bioinformatics,
21(15):3312-3.
5.
Hiller M., Huse K., Platzer M. and Backofen
R. (2005) Non-EST based prediction of exon skipping and intron
retention events using Pfam information. Nucleic
Acids Research, 33(17): 5611-21.
6. Siebert
S. and Backofen R. (2005) MARNA: multiple
alignment and consensus structure prediction of RNAs based on sequence
structure comparisons. Bioinformatics,
21(16):3352-9.
7. Pudimat
R., E.G. Schukat-Talamazzini E.G., and Backofen R. (2005) A multiple feature framework for modelling and predicting transcription
factor binding sites. Bioinformatics,
21(14):3082-8.
8.
Hiller M., Huse K.,
Szafranski K., Jahn N, Hampe J., Schreiber S., Backofen R., and Platzer M
(2004) Widespread occurrence of
alternative splicing at NAGNAG acceptors contributes to proteome plasticity.
Nat Genet. 36(12), 1255-7.
9.
Backofen R., Will S. (2004) Local sequence-structure motifs in RNA. JBCB 2(4), 681 - 698
10. Hiller M., Backofen R., Heymann
S., Busch A., Glaesser T.M., Freytag J.C.. (2004) Efficient prediction of alternative splice
forms using protein domain homology. In
Silico Biology, 4(2), 0017
11.
Backofen R. and Siebert S. Fast detection of common sequence structure patterns
in RNAs. In Symposium on
String Processing and Information Retrieval 2004 (SPIRE 2004).
12. Backofen R. and Busch A. Computational design of new and recombinant
selenoproteins. In Proc. of the 15th Annual Symposium on
Combinatorial Pattern Matching (CPM2004).
13. Backofen
R. and Will S. (2003) A constraint-based
approach to structure prediction for simplified protein models that outperforms
other existing methods. In Proceedings of the 19th
International Conference on Logic Programming (ICLP 2003), pages 49-1.
14. Backofen R. (2004) A polynomial
time upper bound for the number of contacts in the HP-model on the
face-centered-cubic lattice (FCC). Journal
of Discrete Algorithms, 2(2), 161-206
15. Backofen
R. and Sebastian Will S. (2003). A
constraint-based approach to structureprediction for simplified protein models
that outperforms other existing methods. In Proceedings of the
19th International Conference on Logic Programming (ICLP 2003), pages 49-71, 2003.
"Übersicht über
bewilligte Drittmittelprojekte der Jahre 2002-2005":
·
BMBF (FKZ 0312704K): Stochastic Constraint-based Description of Regulatory Sequences, PI: Rolf Backofen, 2003-2007, € 298.007
·
BMBF (FKZ 031652C),
subproject D6: Population genetic
variability of alternative NAGNAG splice acceptors, PIs: Rolf Backofen and Matthias Platzer, 2005-2007,
€ 98.928 (part Backofen)
·
BMBF (FKZ 031652C), subproject D1A: Integrative analysis of complex networks of gene regulation and signal
transduction in cells from patient with rheumatic diseases. PIs: Rolf Backofen and R. Guthke , 2005-2007, € 51.464 (part Backofen)
·
EU Framework 6,
NEST-2003-Path-1 Contr. No. 12835: Emergent organisation in
complex biomolecular systems (EMBIO), PI: Rolf
Backofen, € 124.473
·
EU Framework 6, Network of
Excellence, Project ref. 506779: REWERSE: Reasoning on the
Web with Rules and Semantics, PI: Rolf Backofen,
2004-2008.
·
DFG Sonderforschungsbereich
(SFB) 604 “Multifunctional Signaling Proteins”: Alternative Splicing as a Modulator for Signal transduction, PI: Rolf Backofen, 1 Postdoc position, 2005-2008.
·
DFG priority program
“Selenoproteins”: Replacing cysteine by
selenocysteine in proteins: an algorithmic, bioinformatic approach, PI: Rolf Backofen, 1 Postdoc position, 2001-2003.
7. Project structure (coordination and
composition of the competencecluster, connections with relevant research
institutions)
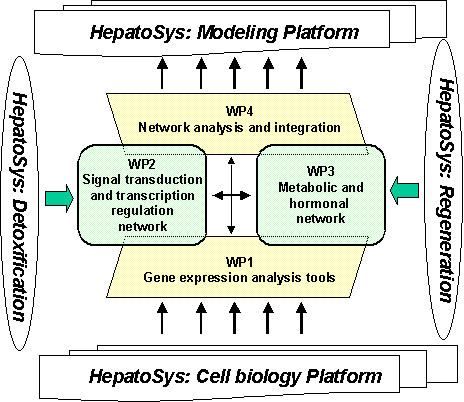
Eight project partners from different
disciplines will cooperate on this project. The HepatoPath project is organized
in 4 Workpackages (WP). The key tasks and interrelationships of the WPs are
depicted in the following schema.
The workflow of
WPs is organized in a working loop, so the results of the work of the
HepatoPath project will be provided as a iteratively enriching platform for all
partners of the HepatoSys research framework.
The partners have
had extensive collaborations, especially in several joint interdisciplinary
research programs founded by BMBF and EU: e.g. Partners 1 and 2 in the BMBF
Bioinformatics Competence Center Braunschweig "Intergenomics" and EU
project "COMBIO"; Partners 1 and 3 and 1 and 4 cooperate on the gene
expression studies of toxicity mechanisms, cancer and cell cycle and other
human disorders.
8. Detailed description
of the work plan and the contribution of each working group
WP1. Gene expression analysis
tools
WP1 will provide bioinformatics software
for the analysis of gene expression and proteomics data. These tools will be
used in WP2 and WP4 for building the gene regulatory networks and
reconstructing of their functional dynamic modes in particular physiological
and pathological states. The tasks are:
1. Development of
novel, high precision methods for predicting TF binding sites in DNA:
multidomain structure of sites, local context feature on flanks, repeated and
symmetrical structure, improving the feature selection algorithms, application
of machine learning techniques, HMMs, genetic algorithms, semi-supervised
learning techniques (Kel, Backofen).
2. Development of
tools for creating promoter models and composite clusters of cis-elements.
Extend the Boolean promoter models to the models of cis-regulatory logic
(promoter programs). (Kel, Wingender, Backofen).
3. Development of a
statistical modeling approach for estimating binding affinity of TFs to their
target sites by learning stochastic models which consider both, sequence data
and quantitative affinity data (Backofen, Kel).
4. Development of
tools for causal analysis of gene expression and proteomics data and
reconstruction of functional gene regulatory networks using integration of
reverse engineering approaches and TF site prediction and phylogenetic
footprinting (Kel, Wingender)
5. Development of
tools for analysis of regulation of post-transcriptional processes, especially
effects of regulated alternative splicing and tools for modelling of regulatory
effects of micro RNAs (Kel, Backofen)
6. Development of an
integrated HepatoPath database for storing of the high throughput
transcriptome, proteome and metabolome data, all generated qualitative and
quantitative data on the regulatory networks of hepatocytes and related cells,
using the the ExProfile database structure as a prototype. (Kel)
WP2. Signal transduction and
transcription regulation network
In WP2 we will experimentally generate and
collect from the literature data on signal transduction and transcription
regulatory networks of hepatocytes in the processes of detoxification,
dedifferentiation, regeneration and related liver pathological states. In WP2
we will create the enriched regulatory networks and deliver them to other WPs
and to the consortia.
1. Updating of the
BIOBASE Knowledge Library (including the databases TRANSFAC, TRANSPATH,
TRANSCompel, HumanPSD and others) with project-relevant signal transduction and
gene regulatory data by manual annotation of the scientific literature and with
the use of text-mining tools. Development of the database structure and
recording of the quantitative regulatory information (Kel).
2. Development of the
novel knowledge-driven ChIP-on-chip method combined with gene expression microarray
analysis. Generation of experimental data on in-vivo genomic targets of several
TFs, important for the hepatocyte regulation, such as, C/EBP, PPAR, HNFs, NF-kappaB, AhR, PXR, ERalpha, p53, AP-1. (Kel,
Borlak, Schmitz, Backofen).
3. High-throughput
transcriptomic and proteomic profiling of the hepatocyte in the processes of
detoxification and regeneration: cell cultures (provided by HepatoSys Cell
Biology platform), different animal models (liver development, liver injury and
regeneration) and primary patient materials (peripheral blood monocytes, plasma
samples and liver biopsy samples from controls, patients with high
triglycerdie/low HDL syndromes (BMI groups), steatosis/NASH patients, and
patients with liver toxicity (cholestasis, alcohol, drug abuse)). (Schmitz,
Borlak, Wuttke, Ramadori).
4. Experimental
studies of signal transduction networks and generation of qualitative and
quantitative data. Analysis of signalling wiring, chaining and kinetic of a
selected set of signal transduction pathways involved in detoxification, cell
cycle, dedifferentiation and regeneration including: MAPK (Ras/Raf/MEK/ERK),
TNF-alpha, Wnt/beta-catenin pathways. (Borlak, Wuttke, Ramadori).
5. Use advanced tools
developed in WP1 to analyze the gene expression data, to identify DNA binding
sites and composite regulatory modules in the promoters of target genes of
transcription factors – master regulators of the processes under study.
In-silico enrichment of the transcription regulatory networks. Validation of
the prediction by various experimental methods (Kel, Wingender, Backofen,
Borlak, Wuttke).
WP3. Metabolic and hormonal
network
In WP3 we will experimentally generate and
collect from the literature quantitative data on enzymes, data on metabolic
pathways and data on network of hormones, growth factors and cytokines that
involve hepatocytes and other cell populations of liver in the processes of
detoxification, dedifferentiation, regeneration and related liver pathologies.
In WP3 we will create the enriched metabolic and hormonal networks and deliver
them to other WPs and to the consortia.
1. Updating of the
BRENDA database with project-relevant data on enzymes and metabolic pathways by
manual annotation of the scientific literature and with the use of text-mining
tools. (Schomburg, Wingender).
2. High-throughput
profiling of metabolome and lipidome of the hepatocyte in the processes of
detoxification and regeneration, using the same biological material as in WP2,
task 4. Storing of the data in the HepatoPath database (Schmitz, Kel)
3. Experimental
studies of hormonal network and generation of qualitative and quantitative
data: a) Animal models of liver injury and regeneration (Ramadori);
b) Analysis of Comparison of effects of selected endocrine disruptors on liver
function with focus on cross-talk of nuclear receptors and gender differences (Wuttke);
4. Updating of the
EndoNet database with project-relevant data on endocrine communication between
different cell types within the liver as well as links between the liver and
the rest of the organism. Further development of the EndoNet structure in order
to enable the collection of quantitative data and representing particular
phenotypes. Populating Endonet with quantitative data (amount, expression,
half-life, affinity, binding constant). (Wingender).
WP4. Network analysis and
integration.
In WP4 we will develop concepts and tools
for integration and structural and analysis of the signal transduction, gene
regulatory, metabolic and hormonal pathways. WP4 will deliver the integrated
comprehensive regulatory network of hepatocytes.
1. Development of the
conceptual schema and of the integration of the four different types of
regulatory pathways, integration of wiring qualitative and quantitative data. (Kel,
Wingender)
2. Further
development of the data exchange formats. Creation of the integrated network of
the hepatocytes and delivery to the HepatoSys Modeling platform (Kel,
Wingender).
3. Development of
tools for network analysis and semi-quantitative modelling (using Petri Nets).
Extend the algorithms to identify key regulators in networks to the weighted
graphs. Incorporation of the information about reaction chaining. Integration
with algorithms of topology and dynamic analysis of networks from other
partners. (Kel, Wingender, Schomburg, Backofen).
4. Case studies:
Application of the constructed integrated regulatory network and network
analysis tools to analyze experimental data in HepatoSys project for
identification of key regulatory circuits involved in detoxification and
regeneration mechanisms in the liver. (Kel, Wingender, Schomburg,
Backofen, Borlak, Schmitz, Wuttke, Ramadori).
9. Detailed costs projection (3 years)
|
|
Personnel
|
Equip-ment |
Consu-mables |
Travel |
Over-head |
Costs |
Requested
Sum |
|
|
BIOBASE |
2 BATIIa |
320000 |
15000 |
2000 |
9000 |
384000 |
730000 |
365000 |
|
UKG-B |
BATIIa+BATVa |
240000 |
5000 |
5000 |
6000 |
24000 |
280000 |
280000 |
|
ITEM |
BATIIa+BATVa |
240000 |
|
135000 |
9000 |
24000 |
408000 |
408000 |
|
UREG |
BATIIa+BATVa |
240000 |
|
45000 |
3000 |
24000 |
312000 |
312000 |
|
IBUC |
BATIIa+BATVa |
240000 |
3000 |
3000 |
4500 |
24000 |
274500 |
274500 |
|
UKG-G |
BATIIa+BATVa |
240000 |
40000 |
60000 |
6000 |
24000 |
370000 |
370000 |
|
UKG-E |
BATIIa+BATVa |
240000 |
|
60000 |
4000 |
24000 |
328000 |
328000 |
|
IIF |
BATIIa+BATVa |
240000 |
|
2000 |
6000 |
24000 |
272000 |
272000 |
|
Total |
9BATIIa
+ 8BATVa |
2.000.000 |
63.000 |
312.000 |
47.500 |
552.000 |
2.974.500 |
2.609.500 |
10. Project time plan
The project duration is considered as
three years. During the first year we will focus on the development of tools
and database structure and establishing the experimental set-up. In the second
year we will generate the main amount of data. During the third year we will
construct the integrated network, analyze the structure and provide it for
further dynamic simulations.
11. Prospects of
success (economic, scientific and/or technical success chances; scientific and
economic utilization chance and continuation capability with dates)
The scientific aim of this project is
ambitious though not unrealistic. First, the problems tackled have to be dealt
with now, since the time is ripe to do so after we entered the
"postgenomics" and "systems biology" era. Second, all
required prerequisites are present: (1) solid data which are properly
structured in databases, knowledge in establishing the still missing
components, and the expertise to integrate and link all these components into an
integrated signaling network; (2) mathematical tools and modeling concept have
been principally established; (3) experiments will be designed to generate all
necessary data and to validate computational predictions. Finally, many of the
project partners including the coordinator have long-standing experience in
many of the related areas and have worked closely together in such an
interdisciplinary setting and even on related projects. The project is
primarily built on existing infrastructure and expertise. These facts should
warrant a successful carrying-out of the project.
The economic value of many of the already
existing bioinformatics tools has been successfully exploited by the industrial
partner BIOBASE, and the expected results of this project may even exceed this
success. No commercially competing initiatives are to be expected since there
are no competitors for the underlying databases. Thus, in case of even partial
success there will be a strong interest to transfer the results into commercial
exploitation, BIOBASE being one candidate to do so.
The achievements
of the project can be flexibly marketed, depending on the specific needs of
potential customers: The commercial partner of the consortium BIOBASE may offer
the products and services resulted from the realization of the project
individually or jointly, since the synergism between the partners will have
been proven at the end of the project. Wherever necessary, individual contracts
will specify the details of the usage of shared IP and exploitation of the IP
of the academic by the commercial partner. E. g., there is already a technology
transfer agreement in place between the partners UKG-G and BIOBASE.
12. Exploitation plan
Exploitation
Strategy
·
Through the industrial partner BIOBASE, as well as through the
technology transfer office of the research centers, we will identify those
technologies, and novel methods that could be patented, as well as possible
partners to licenses them.
·
The partners will reach an agreement on product ownership of the software
produced and on new insight into the networks studied that could lead to new
treatments.
·
The partners will shape the results of the project into altered or
improved marketable products and services. This activity will be done mainly by
industrial partner in the project involving their own marketing and RTD
resources.
Dissemination
and standardization activities
This task will focus on the exploitation
and dissemination of the underlying scientific and technological ideas and
results: by demonstrations and supporting papers at targeted conferences, at
appropriate public events, and of publications in scientific journals of high
quality. Another medium for dissemination will be the presentation on public
WWW servers.